Adeguare gli impianti all’Annex 1: una sfida complessa

Le aziende produttrici di medicinali sterili hanno tempo fino al 25 agosto 2023 per adeguare i propri impianti e produzioni ai nuovi requisiti. Rispetto alla seconda bozza del 2020, la versione finale contiene alcune aggiunte e, soprattutto, molte modifiche del testo con l’obiettivo di renderlo più chiaro e comprensibile.
di Filippo Neri
Il dado ormai è tratto, è scattata la fase operativa di implementazione di tutte le misure necessarie ad adeguare gli impianti di produzione di medicinali sterili ai nuovi requisiti che entreranno in vigore il 25 agosto 2023. In tale data, infatti, avrà termine il periodo transitorio di un anno concesso a partire dalla pubblicazione della versione finale del nuovo Annex 1 alle Good manufacturing practices (Gmp) da parte della Commissione europea.
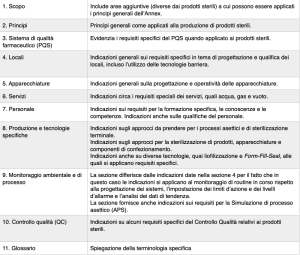
La versione definitiva del documento ha raccolto le ultime indicazioni uscite dalla seconda fase di consultazioni mirate: le cinquantanove pagine totali sono suddivise in undici capitoli, ciascuno dei quali affronta un aspetto specifico delle produzioni sterili (tabella 1). Il documento rappresenta una visione completamente rinnovata di come affrontare lo sviluppo delle produzioni in asepsi rispetto alla precedente versione dell’Annex, risalente al 2008 e che contava solo sedici pagine (box 1).
Due i principi cardine che hanno ispirato la revisione, a partire dalla messa a punto di una Strategia di controllo delle contaminazioni (Contamination control strategy, CCS) sulla base di un’accurata applicazione dei principi del Quality risk management (QRM) a tutte le fasi del ciclo produttivo. Il nuovo Annex 1 ha anche cercato di raccogliere al suo interno tutte le opportunità offerte dall’avanzamento tecnologico analitico e di processo, che si è fatto sempre più veloce e innovativo nel corso del secondo decennio degli anni 2000, alla luce dei requisiti dettati dalle linee guida ICH Q9 (QRM) e Q10 (Pharmaceutical quality system).
Tabella 1 – I contenuti del nuovo Annex 1
(fonte: Commissione europea, Guidelines “The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use”, C(2022) 5938 final del 22.8.2022)
BOX 1 – Storia dell’Annex 1
- 1989: prima versione dell’Annex 1 alle Gmp;
- 30 maggio 2003: prima revisione parziale, in vigore da settembre 2003;
- 25 novembre 2008: seconda revisione parziale, in vigore dal 1° marzo 2009, mirata ad allineare la tabella di classificazione delle camere sterili, includere indicazioni sulle simulazioni dei terreni (media simulations), il monitoraggio del bioburden e la tappatura delle fiale;
- 20 dicembre 2017: pubblicazione della prima bozza di revisione completa, e avvio della prima fase di consultazione pubblica, che ha raccolto oltre 6 mila commenti;
- 20 febbraio 2020: pubblicazione della seconda bozza di revisione completa e avvio della seconda fase di consultazione, che ha coinvolto una gruppo selezionato di sedici associazioni di categoria
- 25 agosto 2022: pubblicazione della versione finale del nuovo Annex 1.
Le principali novità rispetto alla versione del 2020
La versione finale dell’Annex 1 è leggermente più lunga della seconda bozza pubblicata nel 2020 (cinquantadue pagine). Un file di comparazione dei due documenti pubblicato sul sito della ECA Academy (https://www.gmp-compliance.org/files/eca/userFiles/publications/CompareReport%2020220825_gmp-an1_en.pdf) indica un totale di 1.362 modifiche nella versione finale rispetto alla seconda bozza; più in particolare, il testo è stato modificato in 949 occasioni (in giallo), spesso a livello di singole parole; ci sono state anche 263 inserzioni di nuovo testo e 150 cancellazioni (in blu).
A livello dei titoli dei capitoli, è stato tolto il riferimento alle particelle vitali e non vitali da quello del capito 9 (Environmental and process monitoring). È stato anche meglio specificato che i requisiti dell’Annex 1 si applicano “alla progettazione e controllo degli impianti, apparecchiature, sistemi e procedure utilizzati per la produzione di tutti i prodotti sterili”, con il fine di prevenire la contaminazione del prodotto finale da parte di microbi, particolato, endotossine o pirogeni. Non solo farmaci, dunque: le linee guida potrebbero venire utilizzate, ad esempio, anche per supportare la produzione di dispositivi medici o altri prodotti fabbricati in asepsi.
Tra i principi base elencati nel capitolo 2 spicca l’indicazione che impianti, apparecchiature e processi dovrebbero venire “progettati, qualificati e/o validati in modo appropriato e, ove applicabile, soggetti a verifica continua”. Tra le tecnologie utili a tal fine, la versione finale riserva una sezione specifica a isolatori e Rabs (Restricted access barrier system; si veda di seguito); si parla anche di metodi di analisi rapidi/alternativi e continui, in sostituzione dei test rapidi microbici citati nella versione precedente. Un nuovo requisito evidenzia l’opportunità di controllare e testare in modo adeguato anche le materie prime, onde assicurare che il livello di bioburden ed endotossine/pirogeni sia adatto all’uso desiderato. È stata migliorata anche la descrizione degli obiettivi che dovrebbe porsi la Contamination control strategy, che dovrebbe sempre essere supportata da tutta la documentazione utilizzata per svilupparla. Evidenze documentali (come ad esempio i contratti per i servizi di sterilizzazione) dovrebbero essere fornite anche con riferimento ai servizi critici appaltati all’esterno. La CCS dovrebbe sempre comprendere anche la discussione della validazione dei processi di sterilizzazione.

Il capitolo 4 dell’Annex 1 contiene una sezione specifica dedicata alle tecnologie barriera. Viene specificato che isolatori e Rabs sono tecnologie differenti tra loro, utilizzabili per separare gli ambienti di grado A da quelli circostanti (foto Blue Thunder Technologies)
Il design delle camere sterili
Nel capitolo 3 sul Pharmaceutical Quality System è stato introdotto un richiamo specifico al Senior management nel supervisionare lo stato di controllo dei processi, a livello di tutti gli impianti e del ciclo di vita del prodotto (3.1.v). È stato anche specificato che la persona responsabile per il rilascio della certificazione dei prodotti sterili (la Qualified Person nel caso dei farmaci) debba disporre di accesso appropriato a tutta la documentazione di produzione e di qualità.
Il capitolo 4 sui Locali (Premises) dettaglia meglio i diversi gradi di classificazione delle camere sterili (tabella 2), che dovrebbero essere realizzate con materiali che minimizzino la formazione di particolato e che permettano la sanitizzazione ripetuta (4.7). La versione finale dell’Annex 1 specifica che la rimozione di tutti i materiali dalle aree di grado A e B dovrebbe avvenire mediante processo separato unidirezionale, giustificando il ricorso ad altre modalità ove non possibile. Con riferimento agli airlocks, la loro classificazione a riposo è stata ricondotta alle particelle vitali e a quelle totali (e non più vitali e non vitali); lo stesso dicasi per la tabella di classificazione delle camere sterili, i cui valori fanno ora riferimento alla concentrazione massima permessa di particelle totali (4.27, tabella 3).
Altre aggiunte riguardano i requisiti applicabili agli studi di visualizzazione dei flussi d’aria unidirezionali (4.15) e all’opportunità di condurre una riqualifica nel caso d’interruzione del movimento dell’aria che influisca sul funzionamento dell’impianto (4.32.i).
Tabella 2 – La classificazione delle camere sterili
(fonte: Commissione europea, Guidelines “The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use”, C(2022) 5938 final del 22.8.2022)

Le tecnologie barriera
Il capitolo 4 dell’Annex 1 contiene una sezione specifica dedicata alle tecnologie barriera. Viene specificato che isolatori e Rabs sono tecnologie differenti tra loro, utilizzabili per separare gli ambienti di grado A da quelli circostanti. I requisiti sono elencati in modo separato per le due tecnologie (4.19), come pure quelli dell’ambiente di background che le circonda (4.20), di modo da rendere più chiare le attese che dovrebbero trovare riscontro nella progettazione degli impianti.
Il nuovo testo sottolinea anche l’opportunità di ricorrere a tecnologie di trasferimento a elevata capacità o a sistemi validati per l’introduzione dei diversi materiali all’interno dei sistemi barriera. È stata aggiunta l’indicazione che la frequenza di sostituzione dei guanti utilizzati per accedere all’interno di isolatori e Rabs dovrebbe essere definita all’interno della CCS (4.21).
La sezione dedicata alla qualifica delle camere sterili e delle apparecchiature che forniscono l’aria pulita vede l’aggiunta di una frase indicante che “appropriati livelli di pulizia dovrebbero essere mantenuti sia nelle condizioni ‘a riposo’ che nello stato ‘operativo’” (4.23). È stato anche meglio specificato il riferimento alla famiglia di norme ISO 14644 per tutte le attività di qualifica delle camere sterili e delle apparecchiature per l’aria pulita.
La sezione dedicata ai processi di cleaning chiarisce che il programma di sanitizzazione dovrebbe prevedere l’effettiva rimozione dei residui di disinfettanti (4.33). L’utilizzo di quest’ultimi dovrebbe venire validato con riferimento al materiale specifico della superficie, o a un materiale di riferimento previa giustificazione (4.34).
Apparecchiature, servizi e personale
Il capitolo 5 specifica meglio che le parti delle apparecchiature a contatto indiretto col prodotto (anch’esse da sterilizzare) sono quelle che potrebbero entrare a contatto con altre superfici sterili (non con il prodotto), superfici la cui sterilità è comunque critica per quella complessiva (5.5).
Il capitolo 6 discute i Servizi (Utilities). Tra questi, la sezione dedicata ai sistemi per la produzione dell’acqua fa ora riferimento a tutte le fasi di progettazione, costruzione, installazione, messa in funzione, qualifica, monitoraggio e manutenzione degli impianti di trattamento e distribuzione dell’acqua (6.7). Tra i chiarimenti, l’indicazione che la Water for injections (Wfi) può essere prodotta per distillazione o tramite processo equivalente; una possibilità indicata nel testo è l’osmosi inversa accoppiata ad altre tecniche, quali elettrodeionizzazione (Edi), ultrafiltrazione o nanofiltrazione.
Per quanto riguarda il personale, la versione finale dell’Annex specifica che le attività del personale non qualificato dovrebbero essere supervisionate da una persona autorizzata da parte del fabbricante, che dovrebbe anche valutare l’impatto di queste attività sulla pulizia dell’area (7.5). È stata anche aggiunta l’indicazione che le operazioni di vestizione e lavaggio delle mani dovrebbero seguire una procedura specifica per minimizzare il rischio di contaminazione (7.10). Numerose aggiunte e migliorie del testo chiariscono meglio i requisiti applicabili ai materiali per la vestizione e alle procedure per la stessa.
Tabella 3 – Concentrazione massima permessa di particelle totali per la classificazione delle aree sterili
(fonte: Commissione europea, Guidelines “The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use”, C(2022) 5938 final del 22.8.2022)
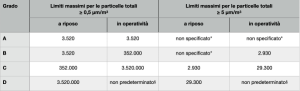
*La classificazione comprensiva delle particelle 5 µm può essere considerata ove indicato dalla CCS o dai trend storici. §Per il grado D, i limiti in operatività non sono predeterminati. Il fabbricante dovrebbe stabilire i limiti in operatività sulla base di una valutazione del rischio e dei dati di routine dove applicabile.
Le tecnologie produttive
Le varie modalità di produzione in asepsi sono discusse all’interno del capitolo 8, che per quanto riguarda le preparazioni e i processi asettici vede l’aggiunta dell’indicazione che ogni intervento o fermo delle linee produttive dovrebbe venire registrato e opportunamente documentato all’interno del batch record (8.17). Tra le attività che dovrebbero venire periodicamente osservate da personale esperto per verificarne la corretta esecuzione è stato inserito anche il comportamento degli operatori all’interno delle camere sterili (8.19).
Le sezioni relative alle tecnologie per la sterilizzazione sono state oggetto di molti interventi per chiarire meglio il testo. Tra questi, si segnala in particolar modo l’introduzione di due sezioni dedicate, rispettivamente, ai processi Form-Fill-Seal (FFS) e Blow-Fill-Seal (BFS); nel secondo caso, sono stati aggiunti alcuni paragrafi che dettagliano meglio le aspettative per questi processi, tra cui gli aspetti che andrebbero considerati nel corso della qualifica dei sistemi (8.114) e i parametri critici di processo (8.115).
Il paragrafo 8.123 sulla sterilizzazione dei liofilizzatori è l’unico che gode di un tempo di transizione più lungo, fino al 25 agosto 2024, sulla base della complessità attesa per l’adeguamento di questo tipo di apparecchiature e processi ai nuovi requisiti. L’indicazione dell’Annex è che, ove i liofilizzatori siano caricati/scaricati manualmente in assenza di sistemi barriera, la sterilizzazione dovrebbe avvenire prima di ogni carico; una dettagliata analisi del rischio dovrebbe rappresentare la base per determinare la frequenza di sterilizzazione nel caso di operazioni automatizzate, da documentare all’interno della CCS.
Il capitolo 9 dedicato al monitoraggio ambientale e di processo ha visto l’aggiunta di alcuni paragrafi, con particolare riguardo alla richiesta di una valutazione di rischio per individuare posizionamento, tipo e frequenza del monitoraggio del personale che opera in prossimità della zona critica (9.25) e il monitoraggio microbiologico del personale che opera nelle aree di grado A e B (9.26). Altre aggiunte descrivono meglio i requisiti richiesti per l’Aseptic process simulation (APS).
Un’implementazione complessa
Mettere a regime le procedure e gli impianti produttivi per adeguarli ai requisiti del nuovo Annex 1 potrebbe in molti casi rivelarsi un’operazione molto complessa, che potrebbe anche comportare la necessità di costruire stabilimenti ex-novo progettati secondo i nuovi principi.
In ogni caso, il punto da cui partire è sempre la raccolta di tutti i dati e le informazioni sui processi attuali, da utilizzare per la predisposizione della Contamination control strategy. Vengono in questo caso in aiuto molte associazioni professionali e di categoria, che hanno sviluppato delle linee guida interne per indirizzare meglio nella valutazione. È il caso, ad esempio, della linea guida su come sviluppare la CCS della ECA Foundation (https://www.eca-foundation.org/news/expert-task-force-updates-ccs-guideline.html), che contiene al suo interno un esempio di gap analisi e uno schema dei documenti e delle linee guida ufficiali, oltre a un possibile modello di documento.
Un altro esempio di possibile approccio è stato pubblicato in un articolo su Outsourced Pharma a firma di Ryan Murray e Amanda McFarland (https://www.outsourcedpharma.com/doc/a-practical-guide-to-navigate-the-eu-s-revised-gmp-annex-0001), che guida il lettore attraverso dieci passaggi chiave. Tra questi, l’identificazione dei gap e delle azioni conseguenti, che dovrebbero essere priorizzate e comunicate a tutti gli attori coinvolti nel processo. La messa a punto e implementazione del piano di azioni correttive e preventive (CAPA) è il passo successivo, seguito dalla misurazione degli esiti ottenuti rispetto a quanto atteso.
 https://icfed.it/wp-content/uploads/2026/05/METAL_DETACTABLE.png
280
376
Eva De Vecchis
http://icfed.it/wp-content/uploads/2024/04/ICF_LogoTestata_Tavola-disegno-1-1.png
Eva De Vecchis2026-05-12 08:00:002026-05-07 15:47:21Elesa porta a Interpack 2026 i suoi componenti standard per il packaging e il processing industriale
https://icfed.it/wp-content/uploads/2026/05/METAL_DETACTABLE.png
280
376
Eva De Vecchis
http://icfed.it/wp-content/uploads/2024/04/ICF_LogoTestata_Tavola-disegno-1-1.png
Eva De Vecchis2026-05-12 08:00:002026-05-07 15:47:21Elesa porta a Interpack 2026 i suoi componenti standard per il packaging e il processing industriale https://icfed.it/wp-content/uploads/2026/05/Interpack23_FS12068-scaled.jpg
1707
2560
Eva De Vecchis
http://icfed.it/wp-content/uploads/2024/04/ICF_LogoTestata_Tavola-disegno-1-1.png
Eva De Vecchis2026-05-08 08:00:002026-05-08 08:49:13ITP a Interpack 2026: film per il pharma, T-Lid riciclabile e soluzioni conformi al PPWR
https://icfed.it/wp-content/uploads/2026/05/Interpack23_FS12068-scaled.jpg
1707
2560
Eva De Vecchis
http://icfed.it/wp-content/uploads/2024/04/ICF_LogoTestata_Tavola-disegno-1-1.png
Eva De Vecchis2026-05-08 08:00:002026-05-08 08:49:13ITP a Interpack 2026: film per il pharma, T-Lid riciclabile e soluzioni conformi al PPWR https://icfed.it/wp-content/uploads/2026/05/Esempio-di-integrazione-tra-sistemi-di-robotica-e-visione-artificiale.jpg
567
852
Eva De Vecchis
http://icfed.it/wp-content/uploads/2024/04/ICF_LogoTestata_Tavola-disegno-1-1.png
Eva De Vecchis2026-05-07 08:00:002026-05-05 07:15:15Steriline a Interpack 2026: flessibilità, customizzazione e conformità all’Annex 1 per un approccio zero loss
https://icfed.it/wp-content/uploads/2026/05/Esempio-di-integrazione-tra-sistemi-di-robotica-e-visione-artificiale.jpg
567
852
Eva De Vecchis
http://icfed.it/wp-content/uploads/2024/04/ICF_LogoTestata_Tavola-disegno-1-1.png
Eva De Vecchis2026-05-07 08:00:002026-05-05 07:15:15Steriline a Interpack 2026: flessibilità, customizzazione e conformità all’Annex 1 per un approccio zero lossULTIMO NUMERO










ULTIME NOTIZIE